

Back to top
miCORE Resequencing

High-resolution mutation detection across entire genomes.
miCORE resequencing provides accurate insights into SNPs, InDels, and structural variants with Illumina sequencing. Whether for CRISPR off-target validation, strain verification, or comparative genomics, our resequencing service delivers the data you need to study genetic variation at scale.
What You Can Achieve
- Detect inherited or acquired mutations, from SNPs to large structural variations
- Compare genomes across strains, isolates, or individuals
- Verify on-target edits and identify off-target mutations from CRISPR/Cas9 experiments
- Confirm genetic modifications in breeding and cell line studies
- Study genome-wide differences linked to disease or phenotype
Before You Start
To set up your resequencing project effectively, consider:
- Scientific objective – mutation detection, genome comparison, CRISPR validation
- Reference genome quality – completeness and annotation status
- Sequencing depth – coverage needed for SNV and InDel calling
- Read length – optimal for resolution of complex variants
- Sample purity – avoid substantial DNA contamination
Modular Workflow
Choose a full-service package or individual modules. A typical resequencing project includes:
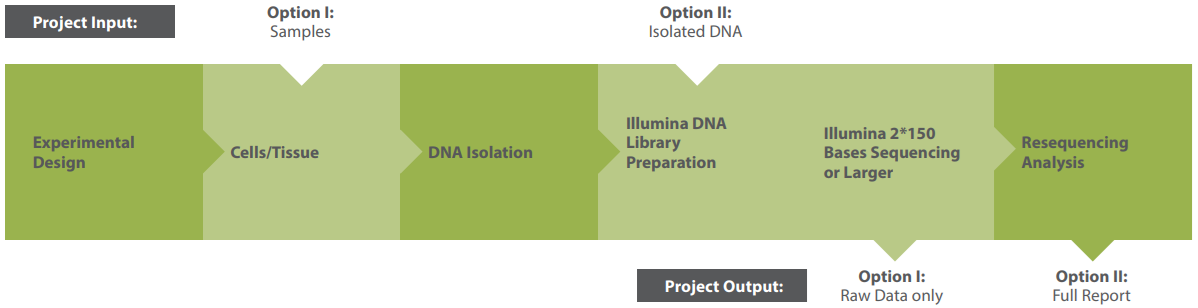
Raw data includes:
- Sequencing QC & yield assessment (.xlsx)
- Raw sequencing reads per sample (.fastq)
- Project summary report (.pdf)
Bioinformatics Analysis
Our resequencing analysis module provides a wide range of deliverables:
- Data quality control
- Alignment and genome coverage analysis
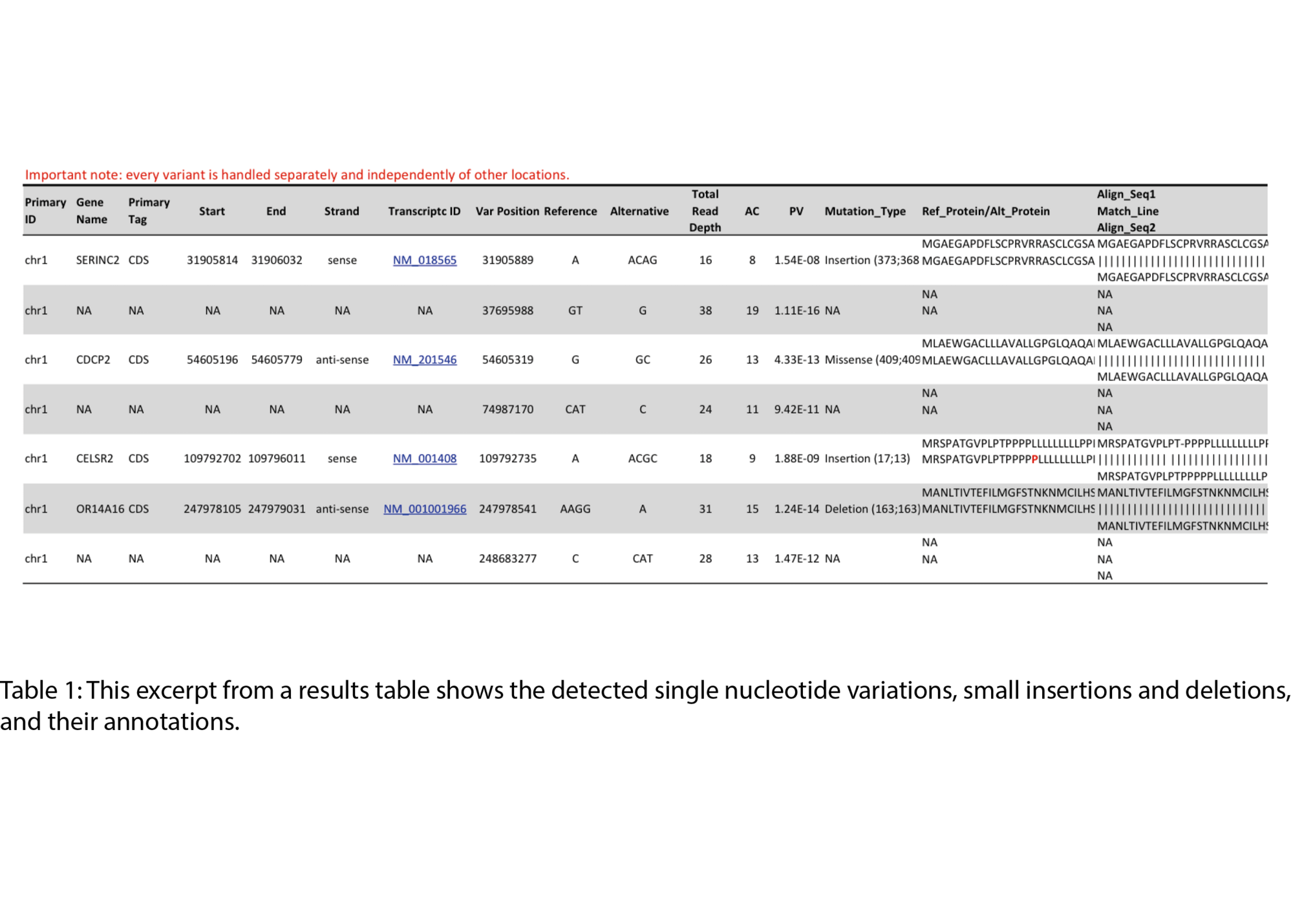
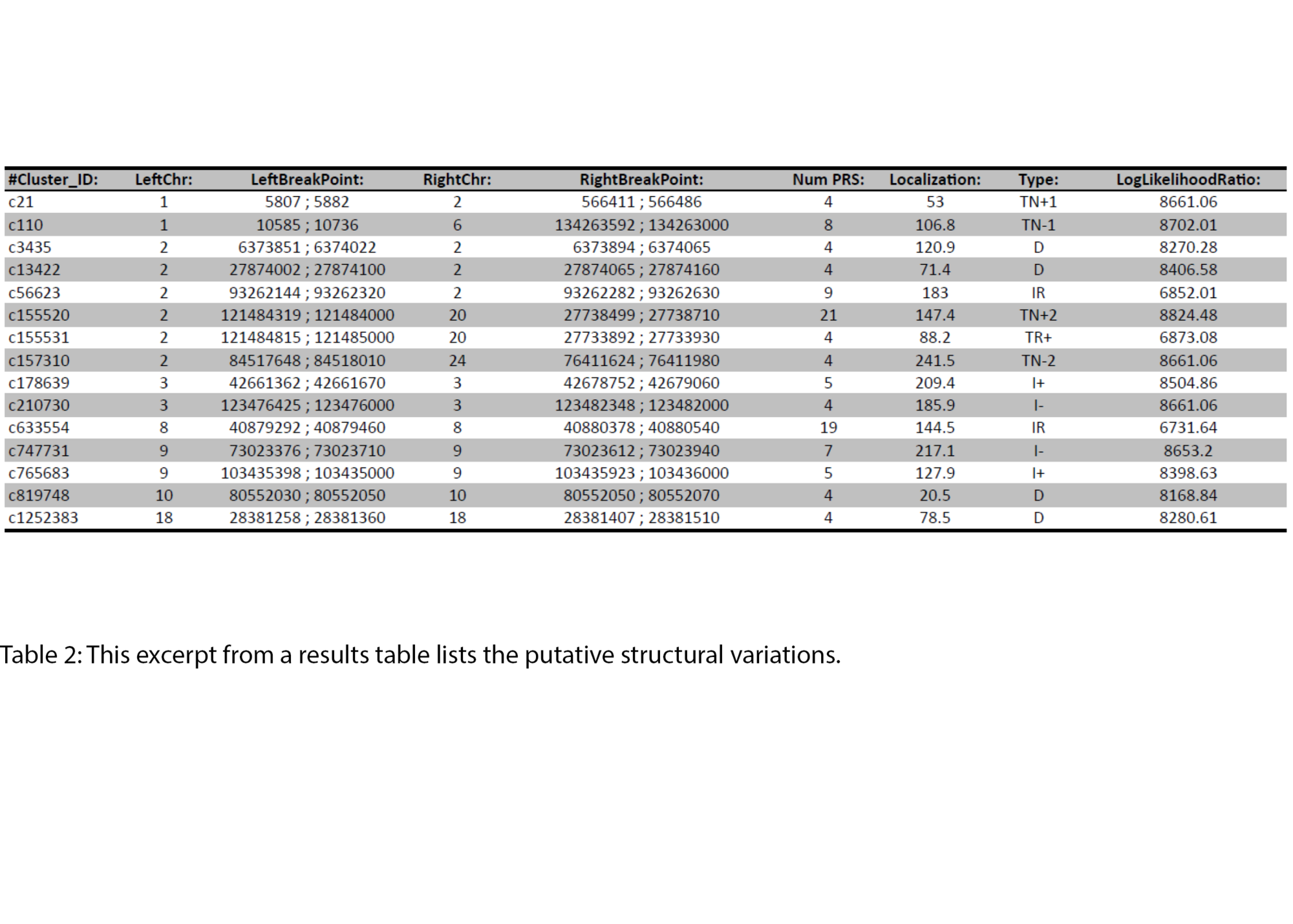
- SNV and small InDel detection (VCF files)
- High-resolution mutation detection across entire genomes
- Annotation of variants with amino acid-level consequences (for annotated references)
- Optional: background filtering, consensus sequence assembly, genomic epidemiology (prokaryotes)
Results are delivered in standard formats (.vcf, .bam, .bed, .html, .pdf) with summary reports for easy interpretation.
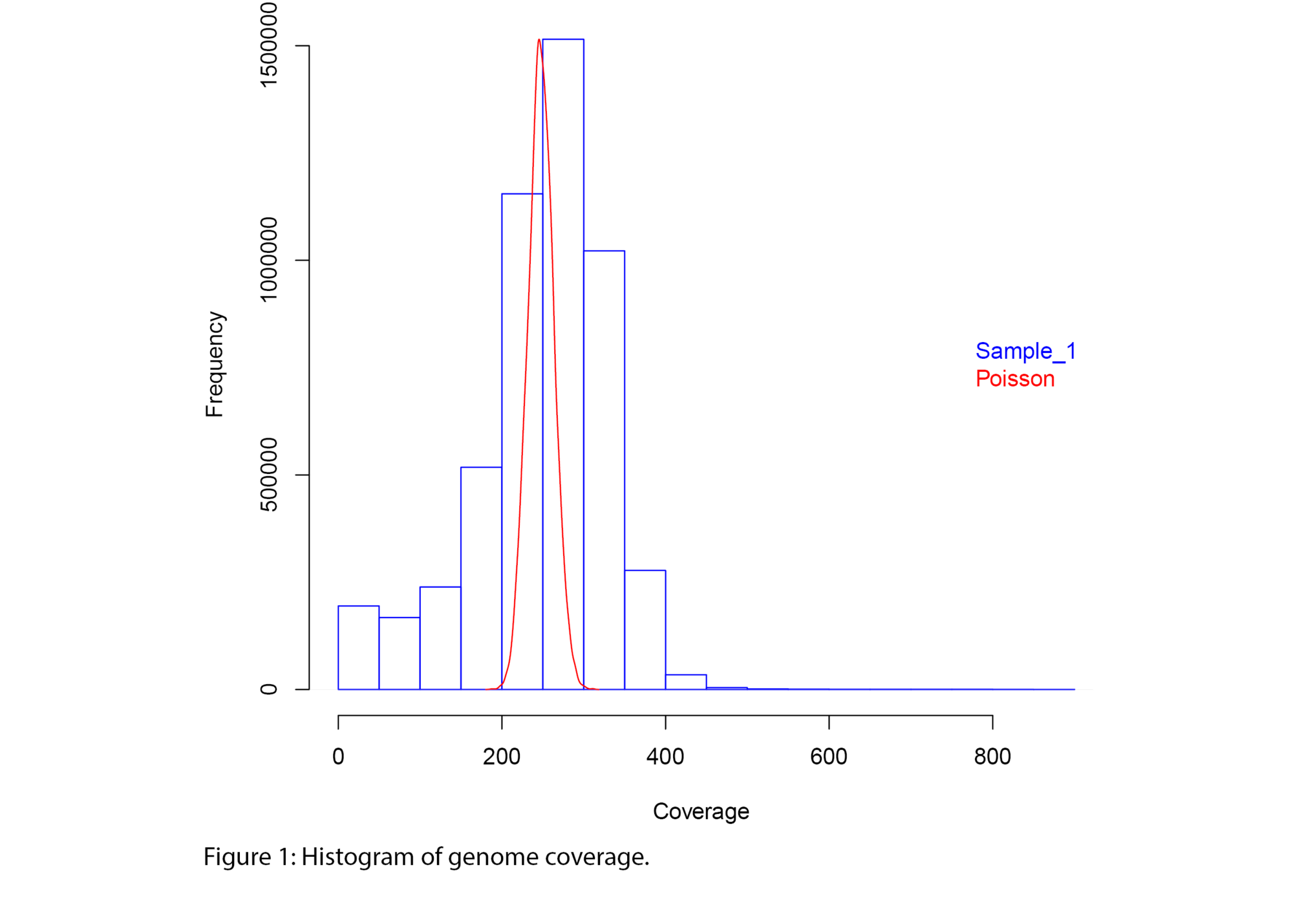
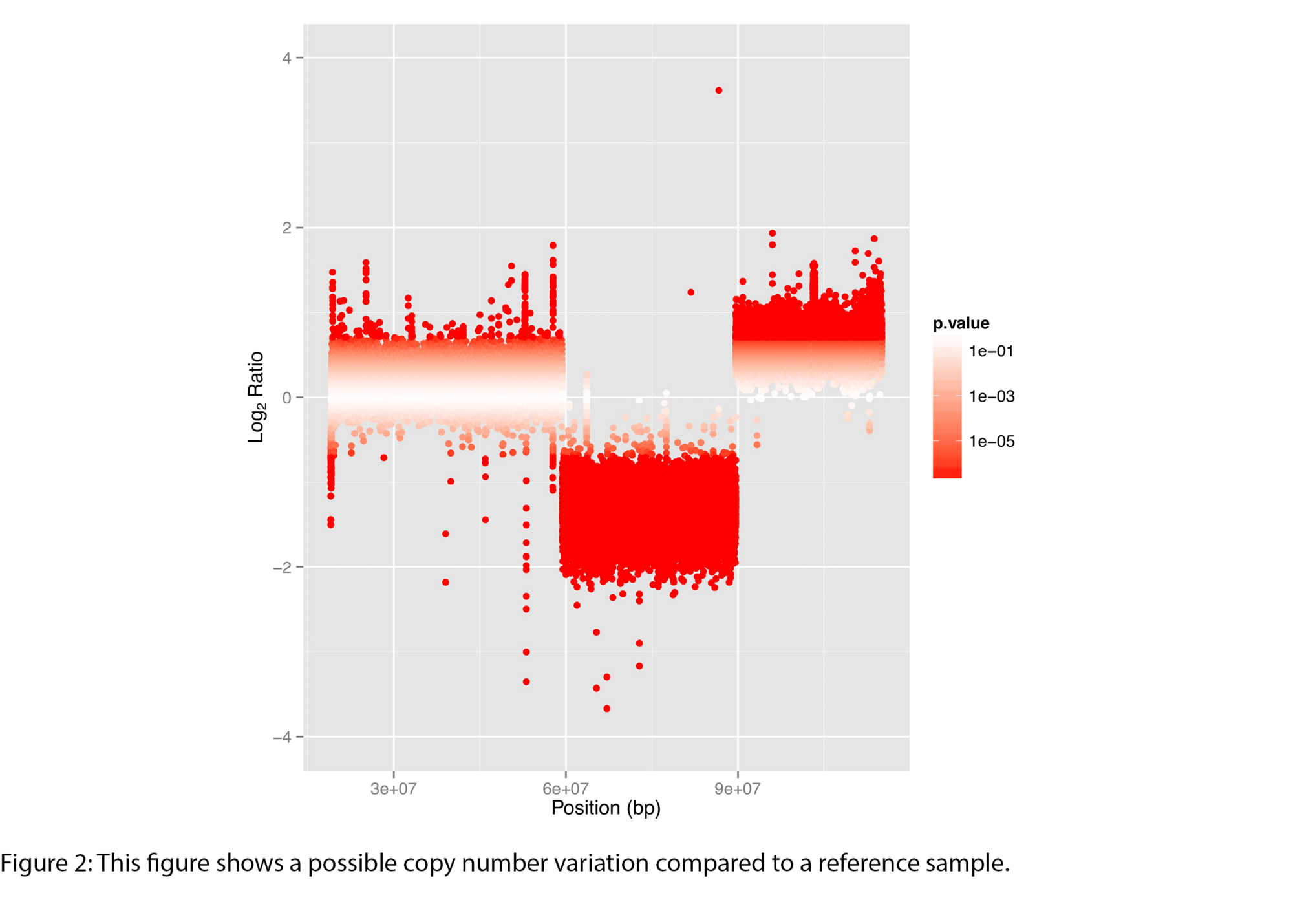
Example Results
All results are delivered in publication-ready figures and interactive formats, providing both high-level overviews and nucleotide-level detail.
Sample Requirements
- See User Guide
Turnaround Time
- Sequencing only: 20 working days
- Data analysis (bioinformatics): +3 working days (small genomes) or +6 working days (large genomes)
- Express service available upon request
Notes
- Related services: Whole Exome Sequencing, miCORE Whole Genome Metagenomics