

Back to top
miCORE mRNA Sequencing

Uncover transcriptome dynamics with precision
mRNA sequencing (RNA-Seq) provides a comprehensive view of gene expression changes under different conditions. From transcript to pathway level, it enables you to analyze differential expression and detect RNA variants with high sensitivity.
What You Can Achieve
- Compare gene expression profiles across experimental conditions
- Identify differentially expressed genes, transcripts, and pathways
- Study alternative splicing and exon usage in eukaryotic transcriptomes
- Detect novel genes and non-coding RNAs
- Explore drug responses, functional changes, and disease mechanisms
- Perform variant calling for SNVs and small InDels in the transcriptome
Before You Start
To design a successful mRNA sequencing project, consider:
- Scientific objective: expression survey, functional study, or variant detection?
- Sample preparation: starting material, RNA fraction (poly(A) enrichment, ribo-depletion)
- Organism & reference: model organism or de novo transcriptome?
- Sequencing depth: sensitivity needed to capture rare transcripts
- Replicates: number required for statistical confidence
- Read length: balance between specificity and cost
- Experimental setup: number of samples, replicates, and conditions
Modular Workflow
A typical workflow for an mRNA sequencing project is shown in the graphic below.
Our modular service design lets you outsource the entire RNA-Seq pipeline or select individual steps
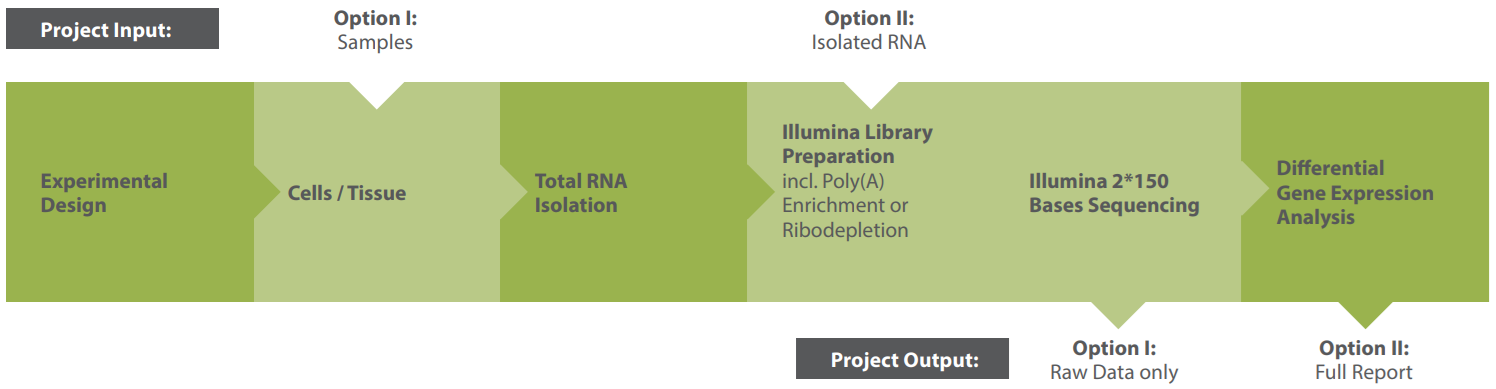
Raw data includes:
- Sequencing QC & yield assessment (.xlsx)
- Raw sequencing reads per sample (.fastq)
- Project summary report (.pdf)
Bioinformatics Analysis
Standard Analysis
Our RNA-Seq bioinformatics module delivers actionable insights:
- Comprehensive interactive report (.html) with sortable and filterable results
- Read alignment & mapping files (.bam/.bai) for complete traceability
- Gene- & transcript-level counts (.tsv)
- Differential gene expression (.tsv & interactive .html)
- Statistical visualizations (heatmaps, volcano plots, enriched pathways, exon maps)
Exclusive to eukaryotic transcriptomes:
- Gene set & pathway enrichment analysis (.tsv & .html)
- Differential exon usage / splicing analysis (.tsv & .html)
Complementary Analysis (Optional)
- Variant calling for SNVs and small (<50 bp) InDels in the transcriptome (.vcf)
This allows combined insights into expression changes and sequence-level variation.
Example Results
Typical outputs include :
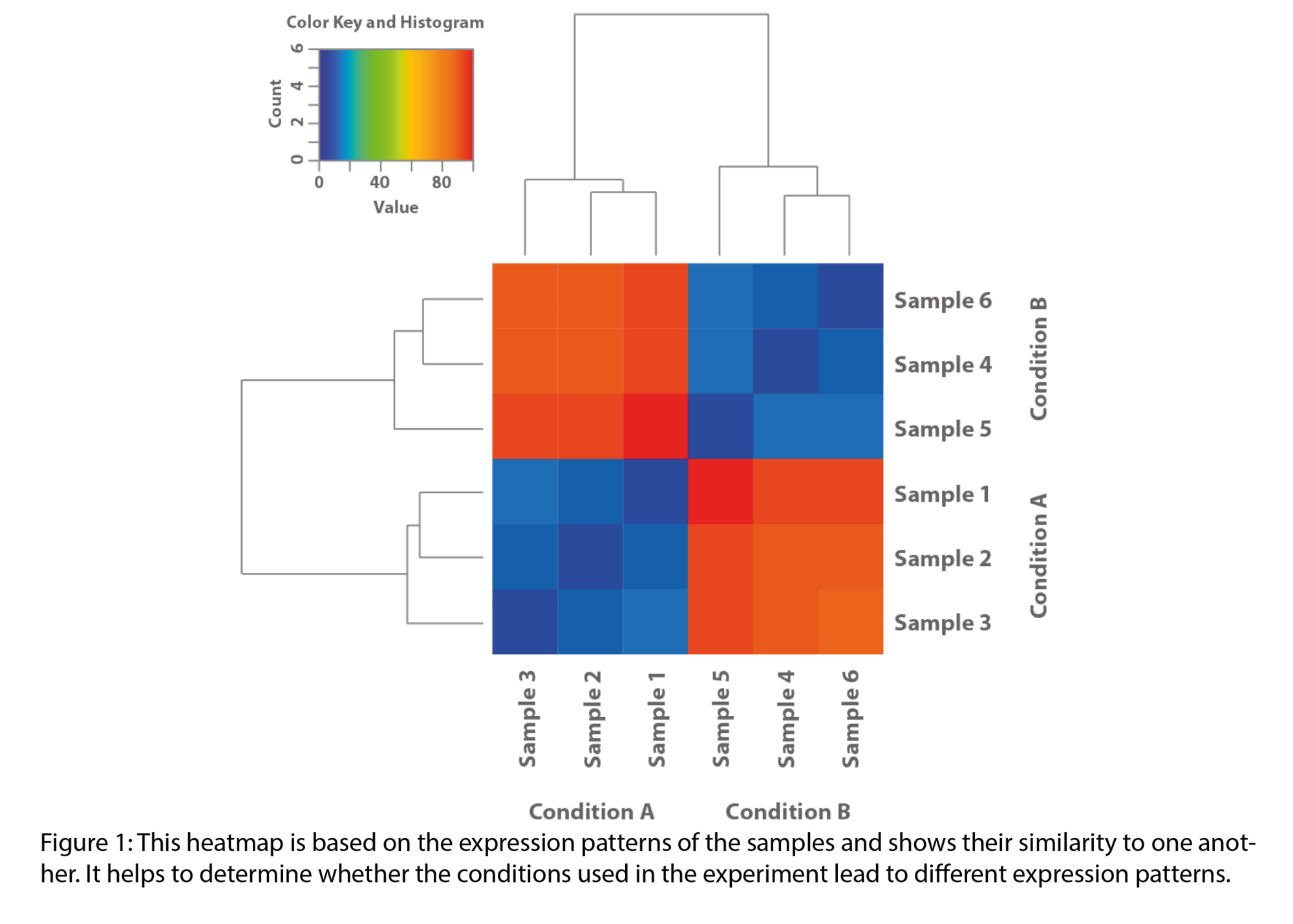
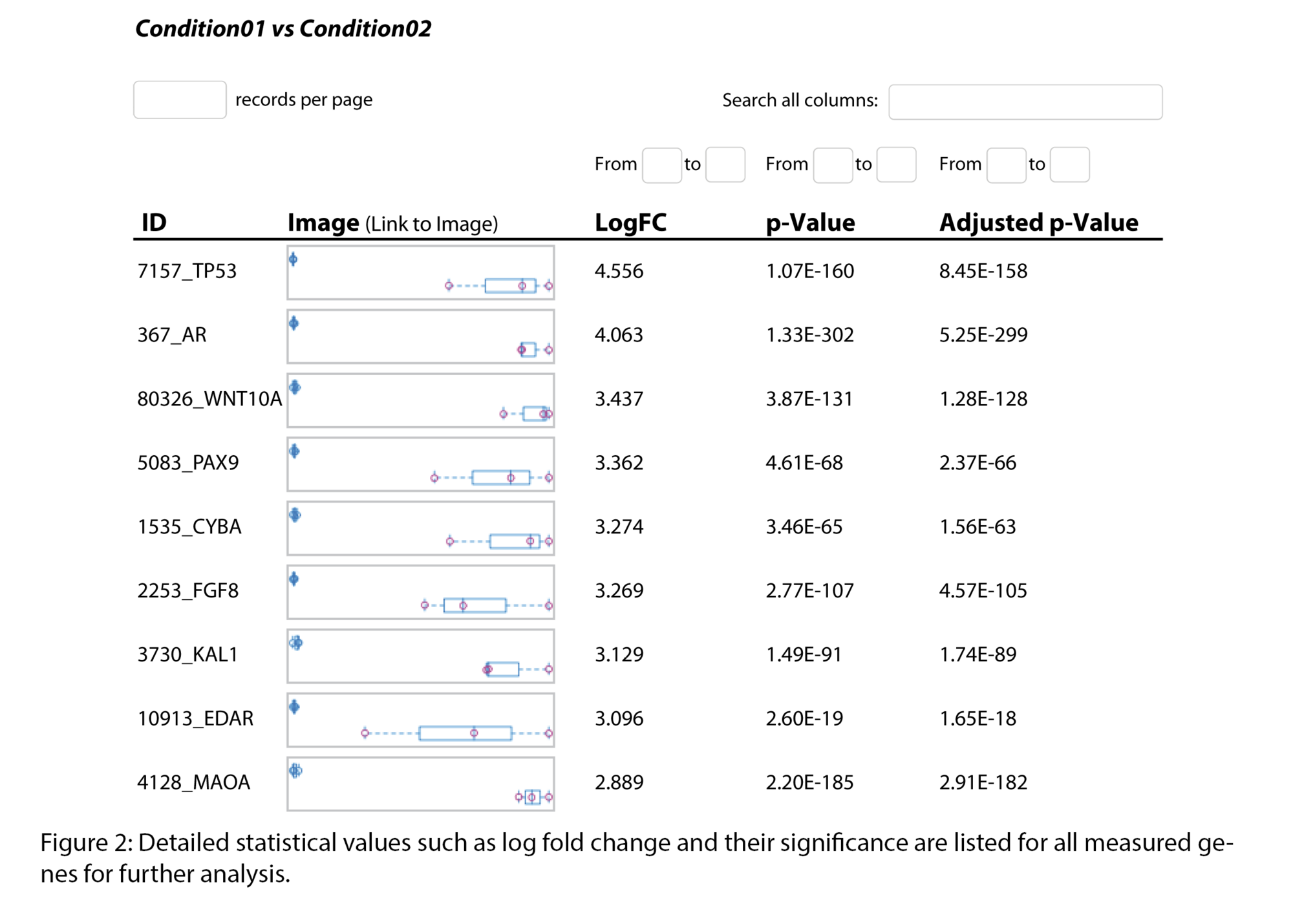
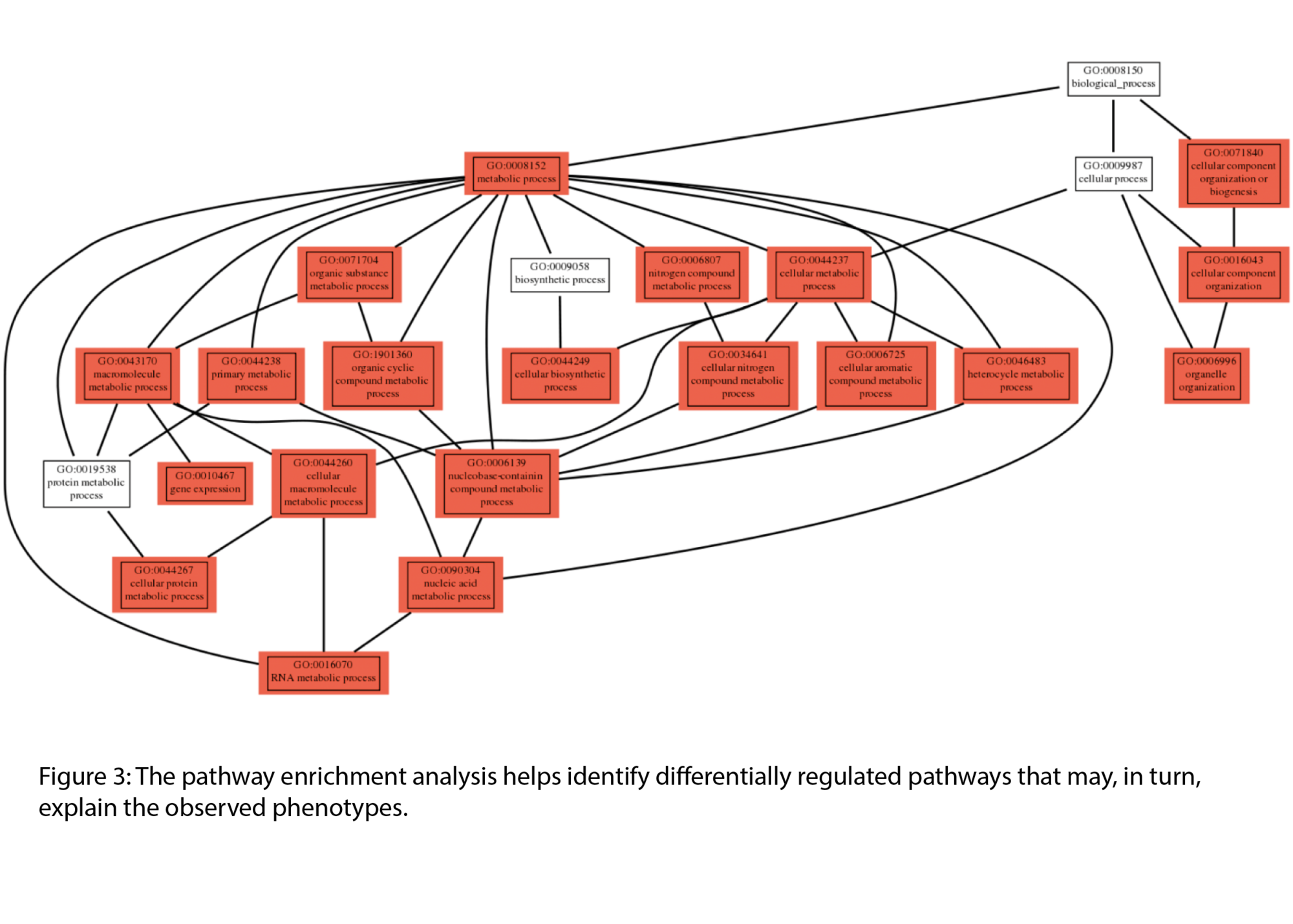
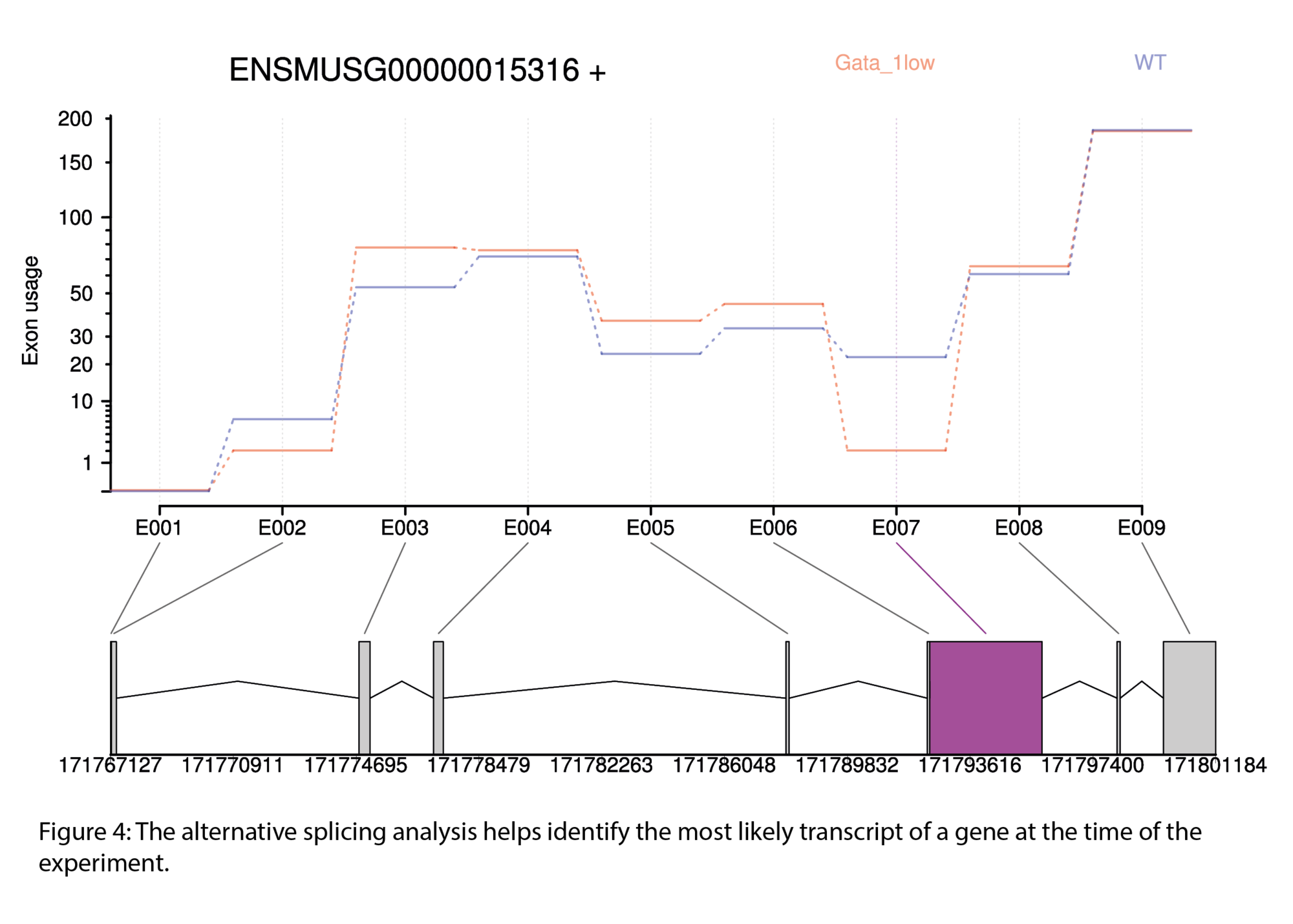
All results are provided as publication-ready figures and, where available, interactive files for deeper exploration.
Sample Requirements
- See User Guide
Turnaround Time
- 15 working days for sequencing (including library preparation)
- RNA isolation: +5 working days
- Full bioinformatics analysis: +3 working days
- Express service available on request
Notes
- Related services: Small and microRNA Sequencing, Shotgun Metatranscriptomics