

Back to top
Amplicon Metagenomics - 16S/ITS

- die taxonomische Zusammensetzung der Gemeinschaft - Bakterien oder Pilze - in Ihren Proben zu untersuchen
- die taxonomischen Verschiebungen innerhalb eines Versuchsaufbaus zu vergleichen
- Ihr nächstes Probenahme-Experiment zu optimieren
Übersicht
Überlegungen vor dem Start eines Amplikon-Metagenomik-Projekts:
- Wissenschaftliches Ziel
- Ausgangsmaterial & gewünschte Dienstleistungen (Isolierung bis Bioinformatik)
- Standard- oder kundenspezifisches Marker- & Primer-Set
- Amplikonlänge (geeignet für Illumina short read Sequencing)
- Probenkomplexität/Sequenzierungstiefe
- Versuchsaufbau (Anzahl der Proben, Wiederholungen, zu vergleichende Bedingungen)
Lassen Sie sich von uns beraten - vom Design bis zur Analyse
Beispielprojekte mit Amplikon-Metagenomik:
- Erkennen Sie nahrungsabhängige Verschiebungen der bakteriellen Darmgemeinschaft
- Untersuchen Sie die genetische Vielfalt von Archaeen in hydrothermalen Tiefsee-Schlot-Umgebungen
- Beobachten Sie Veränderungen der Gemeinschaft in einem Bioreaktoraufbau
- Charakterisieren Sie Pilzsporen aus der Luft
Anwendungen im Zusammenhang mit der Amplikon-Metagenomik:
- Shotgun metagenomics
- Shotgun metatranscriptomics
Workflow

Weitere Informationen sowie eine detaillierte technische Beschreibung finden Sie in unserer Application Note 16S/ITS Metagenomics Sequencing (siehe Download auf der rechten Seite).
Resultate
Ohne Bioinformatik
Rohdaten
Wenn Sie kein Bioinformatik-Analysemodul bestellt haben, bietet Microsynth die folgenden wesentlichen Ergebnisse für 16S/ITS-Metagenomik:
- Bewertung der Quantität und Qualität der Sequenzierung (im .xlsx-Format)
Eine Bewertung der Quantität und Qualität Ihrer Sequenzierungsdaten. - Rohdaten (pro Probe, im .fastq-Format)
Die Rohdaten, die es Ihnen ermöglichen, Ihre eigene Analyse durchzuführen oder jedes sequenzierte Nukleotid zurückzuverfolgen. - Projektzusammenfassung (.pdf-Format)
Ein Bericht, der die wichtigsten Parameter des Projekts zusammenfasst.
Mit Bioinformatik
Standard-Bioinformatik-Analyse
Mit dem Standard-Bioinformatik-Analysemodul von Microsynth für 16S/ITS-Metagenomik erhalten Sie umfassende Einblicke:
- Umfassender Bericht (im interaktiven .html-Format)
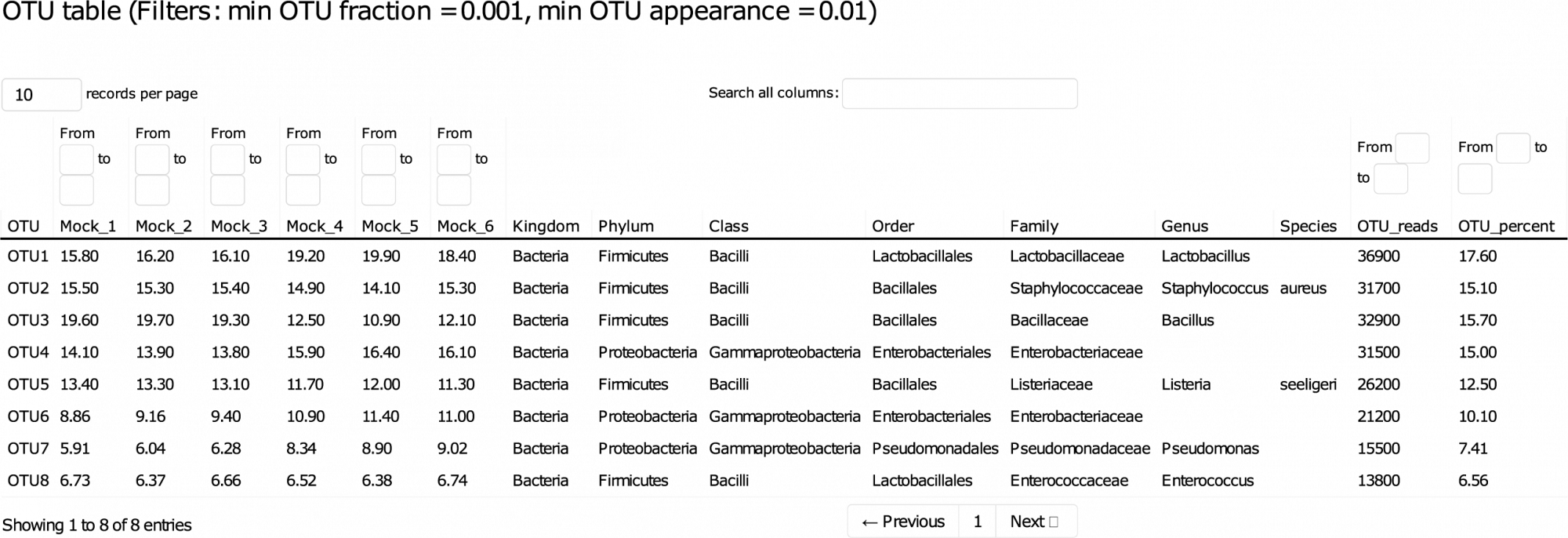
Dieser detaillierte Bericht führt Sie durch die Daten und ermöglicht die interaktive Sortierung und Filterung der wichtigsten Ergebnisse. - Amplikon-Sequenz-Varianten (ASVs) (im .fasta-Format, siehe Tabelle 1)
ASVs, auch bekannt als operative taxonomische Einheiten mit Null-Radius (zOTUs), im .fasta-Format. - ASV-Häufigkeit, Taxonomie und Vorhersagezuverlässigkeit (im .tsv- und .xlsx-Format)
Einblicke in die ASV-Häufigkeit, Taxonomie und Vorhersagesicherheit. - Intrasample Diversity Analysis und Rarefaction Curves (im .tsv und .pdf Format, siehe Abbildung 1)
Erforschen Sie die probeninterne Diversität und schätzen Sie die metagenomische Abdeckung mithilfe von Rarefaction-Kurven. - Metagenom-Visualisierung über Krona Charts (im interaktiven .html-Format, siehe Abbildung 2)
Interpretieren und erforschen Sie Ihre metagenomischen Daten mit Hilfe von interaktiven Krona Charts auf einfache Weise.
Diese Ergebnisse bieten ein umfassendes Verständnis Ihres Experiments, von der Übersicht bis hinunter zur Nukleotidebene, und ermöglichen es Ihnen, fundierte Entscheidungen zu treffen und aussagekräftige Schlussfolgerungen zu ziehen.
Ergänzende bioinformatische Analysen (gegen Aufpreis):
1. Vergleichende Analyse
- Probenübergreifende Diversität (im .tsv- und .pdf-Format, siehe Abbildung 3)
Gewinnen Sie Einblicke in die Diversität zwischen den Proben, dargestellt in tabellarischer und grafischer Form. - Hauptkomponentenanalyse (im .tsv- und .pdf-Format, siehe Abbildung 3)
Visualisieren und interpretieren Sie komplexe Beziehungen durch die Hauptkomponentenanalyse. - Differenzielle ASV-Analyse (im .tsv- und .html-Format, siehe Tabelle 2)
Identifizieren und untersuchen Sie differentielle ASVs mit detaillierten Analyseergebnissen, die sowohl im Tabellen- als auch im HTML-Format verfügbar sind. - Visualisierung der statistischen Analyse
Greifen Sie auf visuell informative Darstellungen von statistischen Analysen zu.
Diese zusätzlichen Dienste bieten eine tiefere Ebene des Verständnisses und der Interpretation und verbessern Ihre Fähigkeit, wertvolle Erkenntnisse aus Ihren Proben zu gewinnen.
2. Funktionelle Profilierung
- Inferenz und Vorhersage von funktionellen Profilen (.tsv- und .html-Format, siehe Tabelle 3)
Prognostizieren Sie das funktionelle genetische Potenzial Ihrer Proben auf der Grundlage von Erhebungen und vollständigen genetischen Informationen der identifizierten Taxa.
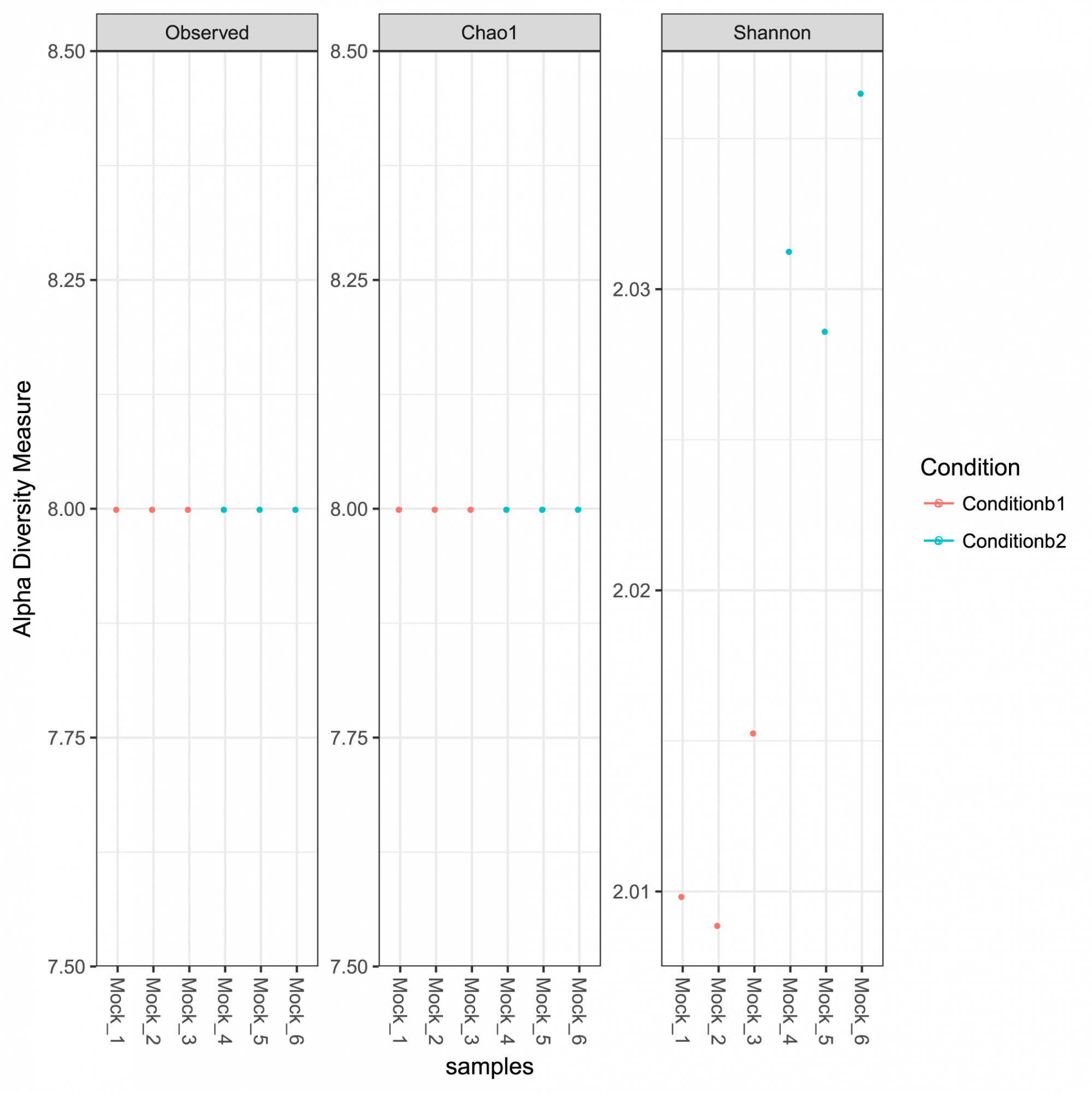
Abbildung 1: Alpha-Diversitätsmaße für die analysierte Gemeinschaft, einschließlich der beobachteten Reichhaltigkeit, Chao-1-Indizes, die die geschätzte Reichhaltigkeit darstellen, und die Shannon-Diversitätsindizes.
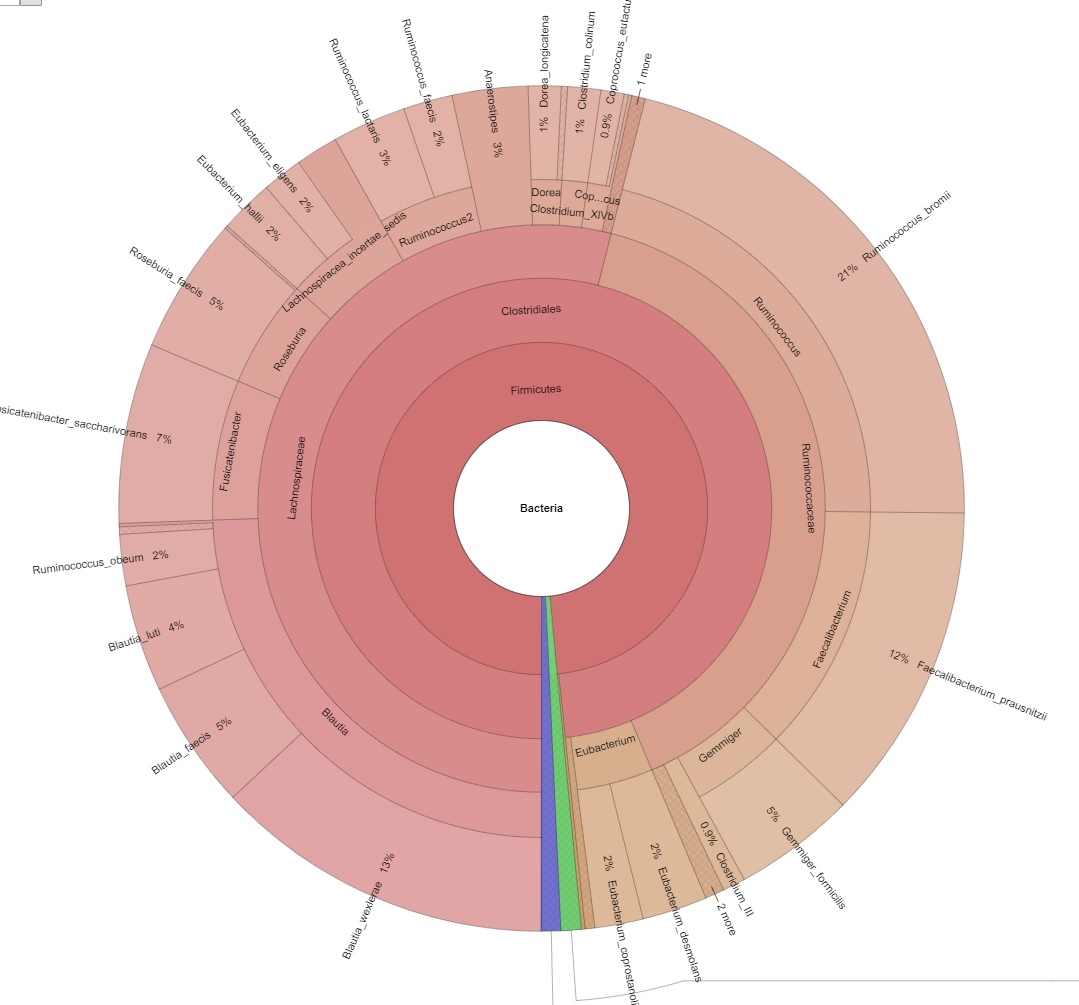
Abbildung 2: Bild eines interaktiven Krona-Charts.
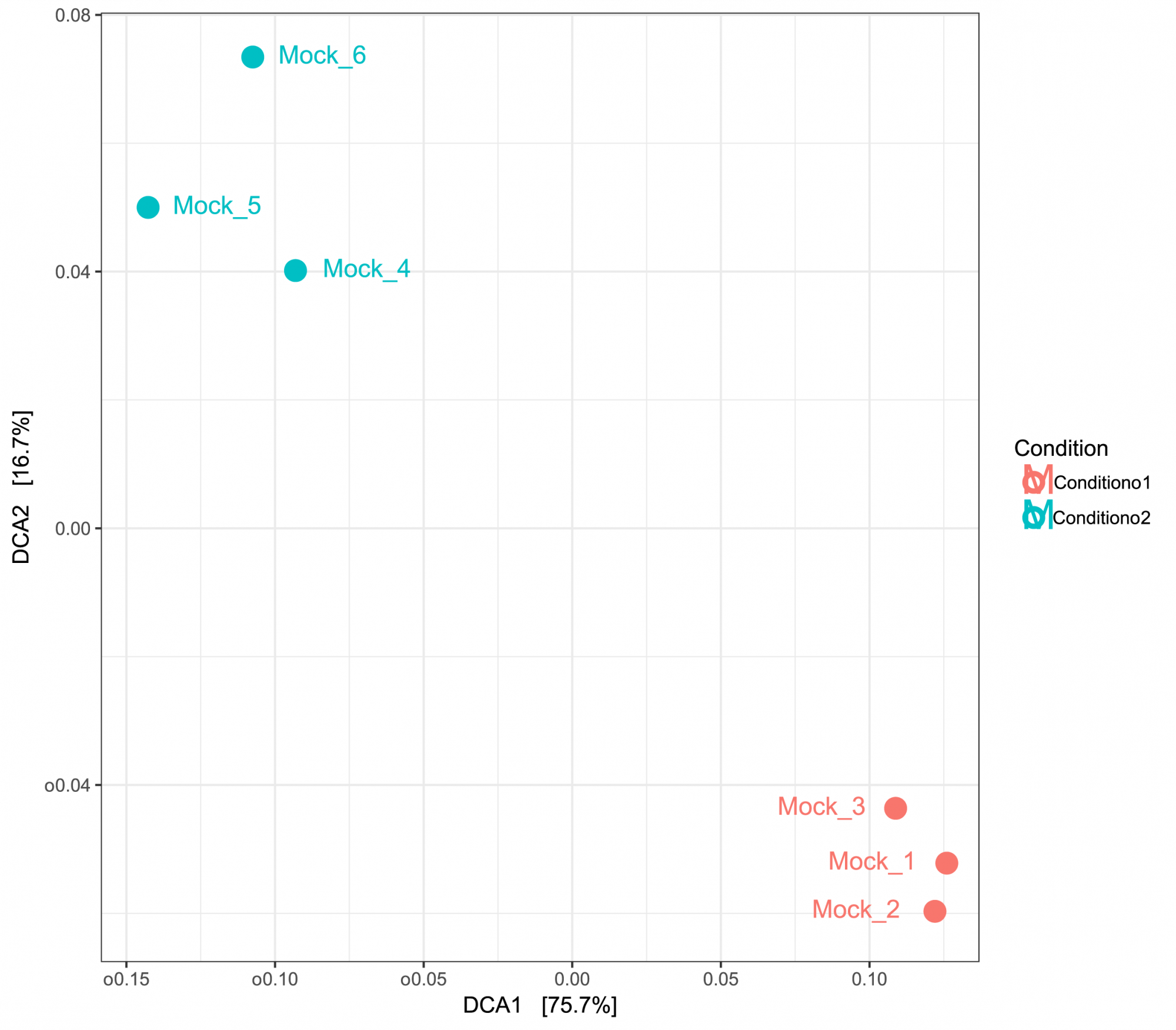
Abbildung 2: PCA basierend auf UniFrac-Distanzen, die Ähnlichkeiten zwischen den Proben anzeigen. Die Proben wurden nach zwei Bedingungen gruppiert.
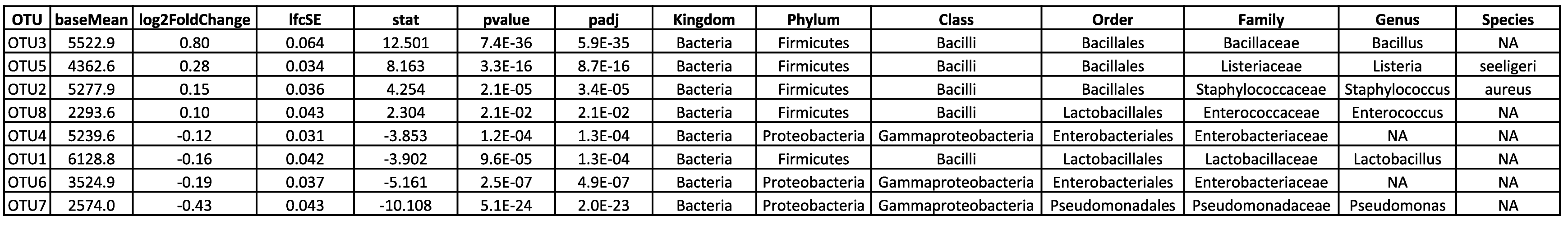
Tabelle 1: Dieser Ausschnitt einer Ergebnistabelle listet die relativen Häufigkeiten und taxonomischen Identifikationen der beobachteten OTUs in den verschiedenen Proben auf. Für die taxonomische Klassifizierung werden Konfidenzwerte berechnet und OTUs werden nur dann in einen bestimmten Rang eingestuft, wenn ihr Konfidenzwert über einem bestimmten Schwellenwert liegt, um zweifelhafte Klassifizierungen und falsch-positive Ergebnisse zu vermeiden.

Bearbeitungszeiten
- Lieferung der Daten innerhalb von 20 Arbeitstagen nach Probeneingang (inklusive Library Erstellung und Sequenzierung)
- Weitere 5 Arbeitstage für die Datenanalyse (Bioinformatik)
- Express-Service auf Anfrage möglich