Whole exome sequencing (WES) or exome sequencing has emerged as a routine resequencing technique for all protein-coding genes in a genome (the exome). Despite many protocols that differ in detail, the principle remains the same. In a first step, the genomic DNA is reduced to only the coding regions of the genes, known as exons (about 180,000 exons for humans). In a second
step, the resulting exonic DNA is sequenced using next generation sequencing (NGS) technology. By focusing on the protein coding regions of the DNA (approximately 1% of the human genome), the identification of the functionally relevant genetic variants that alter protein coding are detected at much lower costs compared to whole genome sequencing (WGS). Therefore, WES is widely used for cancer
exome sequencing, causal variant studies (mono- and polygenic diseases) or translational research. The affordable sequencing depth makes exome sequencing well suited to several applications that are based on reliable variant calls such as clinical diagnostics and rare variant mapping in complex disorders.
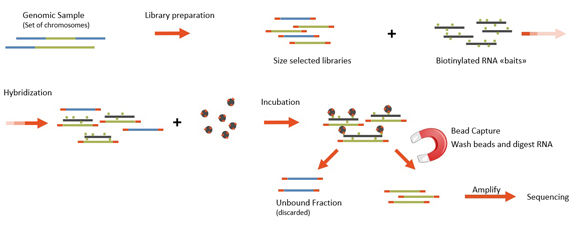
Figure 1: Schematic workflow of exome capture and high throughput sequencing (see section “Enrichment & Sequencing” for details).
Microsynth Competences and Services
Experimental Design Unlike other NGS projects, the setup for a whole exome sequencing project seems rather straightforward. Even replicates are not mandatory in numerous settings. Nevertheless, a thorough consulting by our experts helps you to get the best out of your study.
DNA Isolation
You may either perform the DNA extraction yourself or outsource this critical step to Microsynth. We have long-standing experience in DNA and RNA isolation from demanding matrices including isolation from fixed samples and serum.
Enrichment & Sequencing
The Agilent SureSelect in-solution capture is applied for targeting the human exons. Key benefits of the Human Whole Exome V6 + COSMIC panel are
the comparable deep and complete target coverage, the high “on target” coverage, inclusion of more exons on the hard-to-capture targets and additional coverage of cancer relevant targets with the COSMIC-baits. The RNA-driven DNA capture is conducted by RNA oligonucleotides (or “baits”) that are biotinylated for easy capture onto streptavidin-labeled magnetic beads (see Figure 1). To perform the capture, genomic DNA is sheared and assembled into a library format. Size selection is performed on the library prior to capture. Size-selected libraries are then incubated with the baits, and RNA bait-DNA hybrids are then “fished” out of the complex mixture by incubation with the magnetic beads. RNA
baits are digested such that the remaining nuclear acid is the targeted DNA of interest. Captured DNA is amplified and the targeted samples are sequenced (2x75 bp), resulting in at least a 100-fold coverage per sample.
Bioinformatics Analysis
Our WES Service allows several entry points and processing grades for our deliverables (see Figure 2). The Whole Exome Sequencing analysis consists of reads mapping to the human reference genome, calling and annotation of single nucleotide variations as well as small insertions and deletions (see Figure 3). Results are compared to dbSNP databases (e.g. ClinVar-NCBI-NIH). Additionally, a comprehensive coverage analysis of all captured exons is provided.
Figure 2: This chart depicts a typical example workflow for a whole exome sequencing project at Microsynth and lists all possible input and output points.
Example Results of the Whole Exome Sequencing Analysis
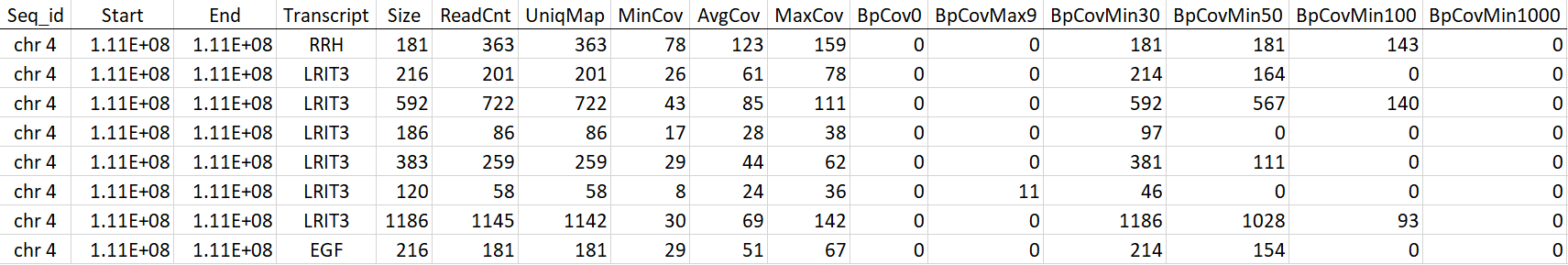
Table 1: This is a cutout of the full table detailing read coverage for every target in the sequenced exome.
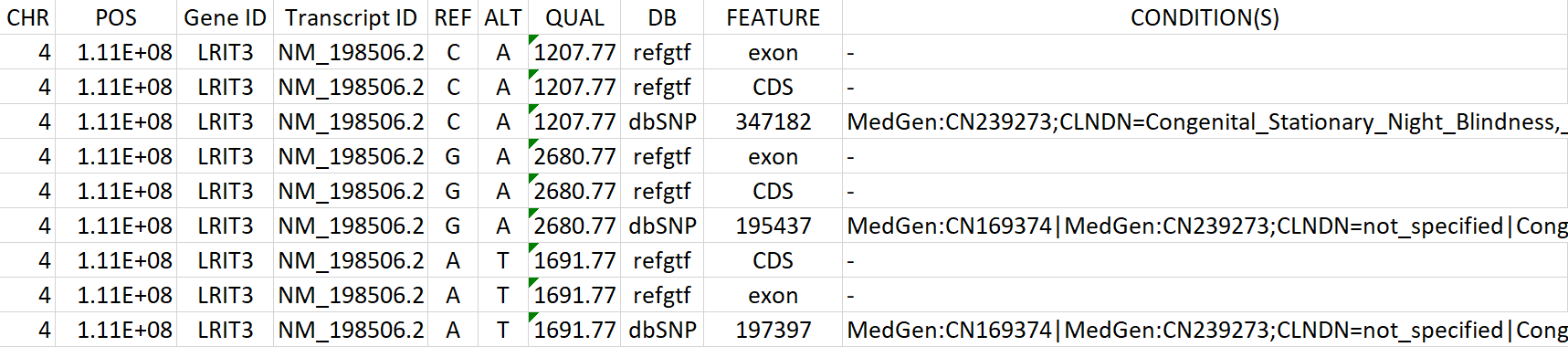
Table 2: This excerpt of a results table shows detected variations (alternative compared to the reference), their annotation and whether they and their impact are already known (e.g. as ClinVar - Variations). (Chromosomal position, gene and transcript IDs, feature type and numerous cross references not depicted in the excerpt)
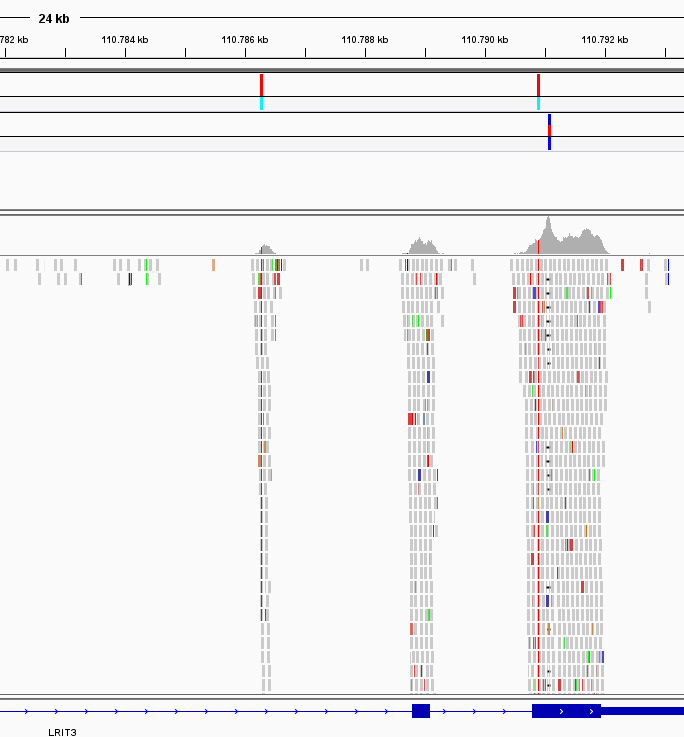
Figure 3: The data allows a detailed data inspection with a genome browser. Besides the refined data, raw and intermediate data in standard file formats are provided. This allows an overarching inspection and in-depth verification of the results by publicly available tools.